← Back
LS-MolGen | 靶点口袋结构和活性分子数据双驱动的人工智能分子生成方法加速药物分子发现
2022年2月
【有关新药研发和AIDD的背景介绍】
新药研发是一个高风险、高投入、高回报,但是研发周期长的多学科交叉技术行业。一个药物从最初的发现到上市,再到进入临床为患者带来生存获益,都要经历大量、严格的数据验证,通常需要花费10~20年时间,投入金额高达5亿~26亿美元。新药研发一般包括5个阶段:药物发现、药物临床前研究、药物临床研究、药品的申报与审批阶段和新药监测阶段,其中药物发现包括药物作用靶点(target)以及生物标记(biomarker)的选择与确认、先导化合物(lead compound)的确定、构效关系的研究与活性化合物的筛选、候选药物(candidate)的选定等过程。因此,先导化合物的确定,对于决定一款药物最终能否成功上市显得至关重要。

新药研发的五个阶段
近年来,深度学习在很多领域得到广泛的应用,其中自然也包括药物发现。特别地,AI在计算机辅助药物设计(CADD)方面取得了突破进展,也称为人工智能辅助药物设计(AIDD)。一项发表在Nature子刊上的工作吸引了很多科学家以及企业界的注意,文章中开发了一个名为GENTRL的深度生成模型,运用它可以在46天内开发出针对盘状结构域受体1激酶(DDR1)的强效抑制剂,其中包括21天的生成分子过程,相比药物发现3~6年的时间,这毫无疑问大大节省了时间成本。
【常见人工智能方法辅助药物分子设计的不足之处与改进方向】
一般来说,针对靶蛋白设计药物的方法可以分为基于配体和基于结构的方法。基于配体的方法,利用已知特定目标的已知活性配体数据集来设计具有最佳特性的有效分子,完全由数据驱动来有效地生成化合物,如图2(A)所示。而基于结构设计化合物分子的方法,则利用靶点的高精度的3D结构特征进行化合物分子生成,在这个过程中,靶点蛋白口袋的3D结构信息为优化配体和靶标之间的相互作用提供直接指导,从而驱动如图2(B)所示的合理化合物设计。然而,基于配体的方法可能会根据训练集中的配体产生偏差,并且在缺少靶点口袋结构的情况下,该方法生成的分子和训练集的分子具有非常高的相似性,不利于开发能够避开专利的全新分子;而单纯基于靶点结构进行分子生成的方法,在缺少已知化合物信息的情况下,可能难以快速收敛到满足性质需求的分子化合物空间。因此,针对一个特定的靶点蛋白开发药物分子,并且该靶点具有一系列已知的有生物活性的配体分子和蛋白质三维晶体结构,开发一种更具包容性的、可以结合基于结构和基于配体的药物设计方法二者优点的新方法,如图2(C)所示,是非常必要的。
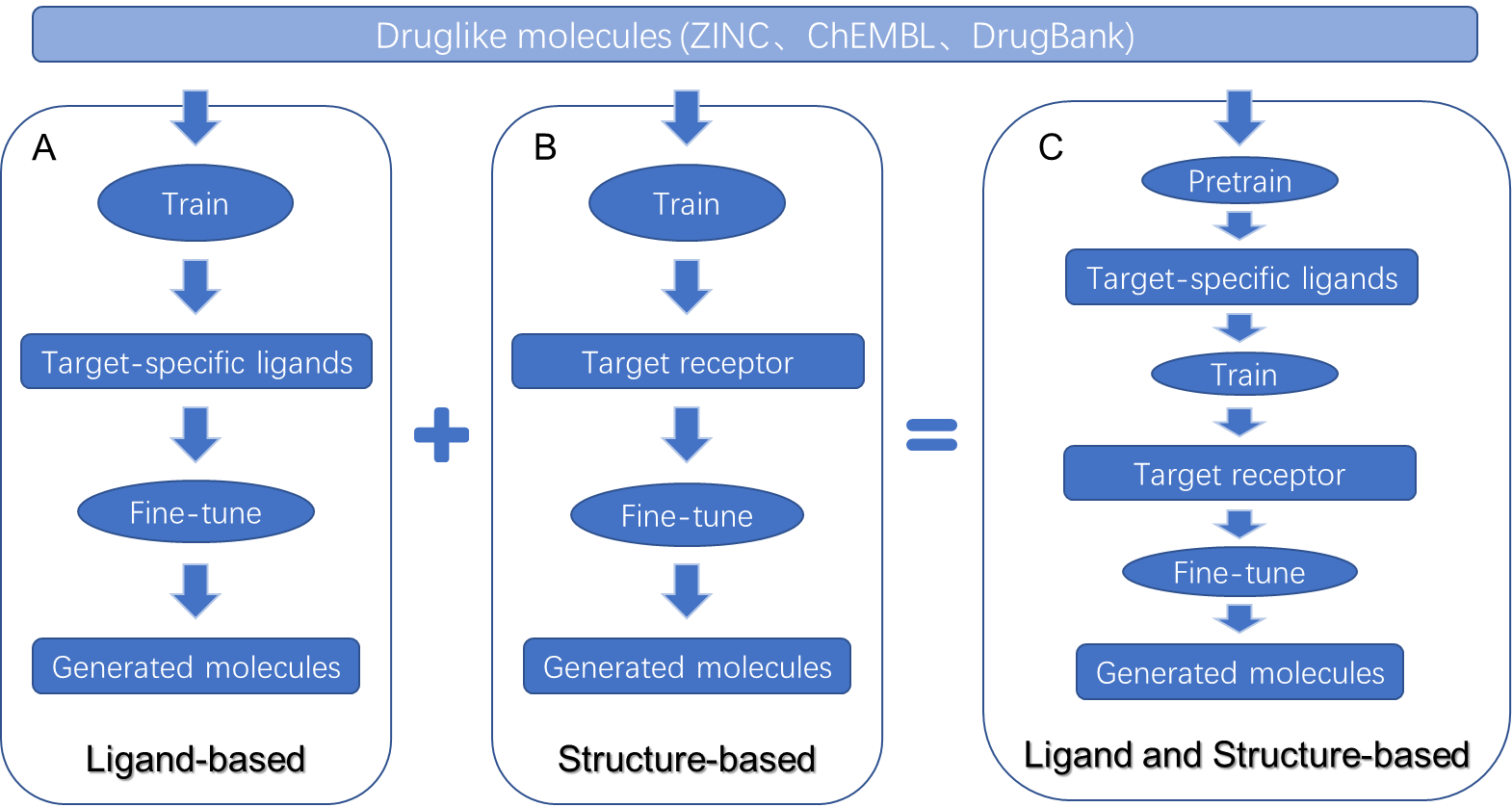
基于活性配体信息构建模型(A)、基于靶点结构信息构建模型(B)、以及同时基于活性配体和靶点结构信息构建模型(C)
的深度学习方法示意图
【结构与数据双驱动的人工智能分子生成方法介绍】
基于此,我们开发了一种可以兼顾利用已知配体和靶点结构信息进行分子生成的方法(LS-MolGen),如图3所示,该方法可以有效的结合前面所提到基于配体生成分子和基于靶点结构生成分子的优势。具体来讲,LS-MolGen首先将公开大数据集(ChEMBL、ZINC等)上的类药分子作为预训练的语料进行一轮大规模的预训练,该过程可以学习到分子SMILES的语法规则以及类药分子的空间分布形式;之后进一步地将大数据集上学习到的知识迁移到针对特定靶点的小数据集,利用该靶点的已知活性小分子配体对模型参数进行调整;最后将模型学习到知识初始化到强化学习的智能体网络中,通过强化学习高效搜索和该靶点亲和力高的分子,搜索的奖励函数设置为小分子和靶点口袋对接的分数。
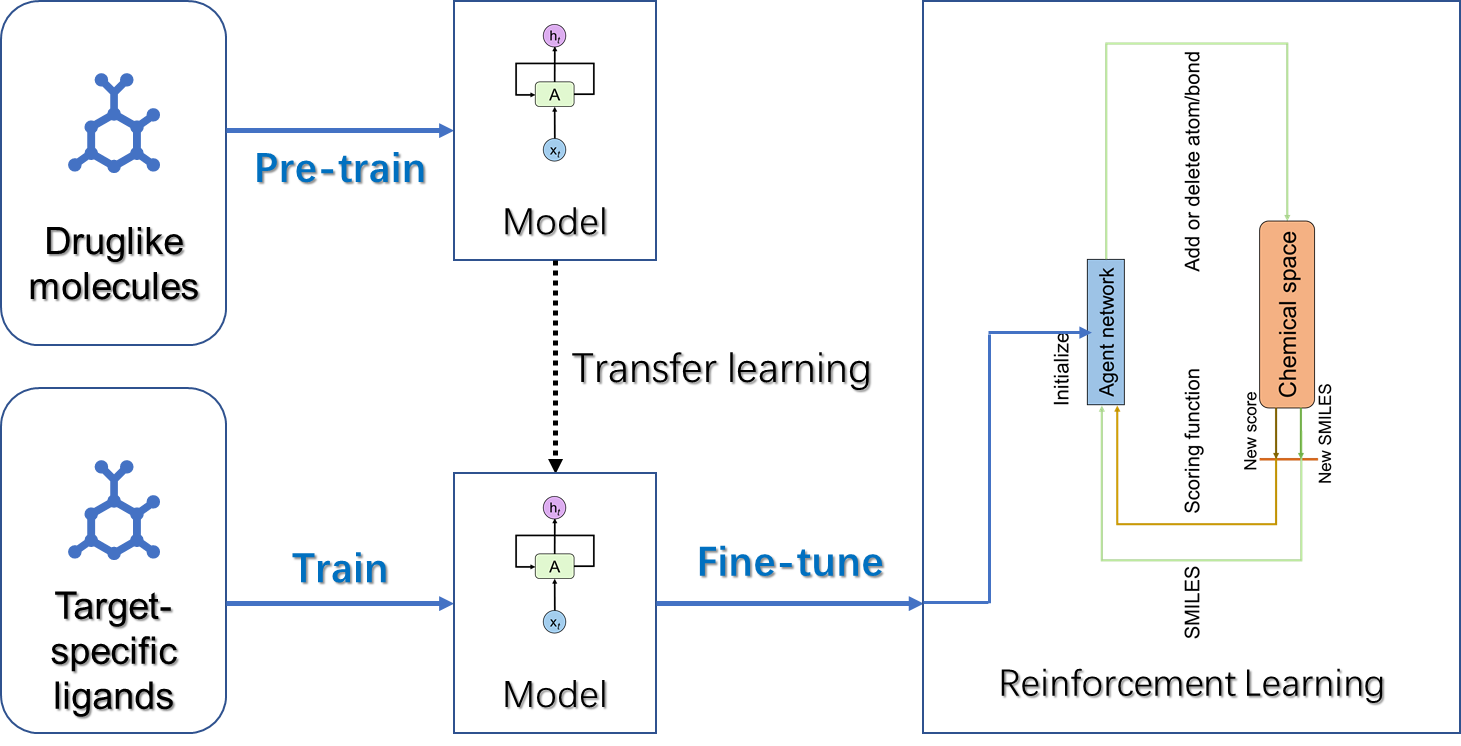
LS-MolGen分子生成方法
【应用LS-MolGen的药物分子开发案例】
LS-MolGen方法可以有效结合配体和靶点结构的信息,生成高质量的全新分子库。以某类GPCR蛋白靶点的拮抗剂的开发为例,我们利用LS-MolGen生成上万级别的全新化合物分子,配合活性预测模型、ADMET性质预测模型、分子对接分析、自由能微扰计算以及专家经验知识评估,我们最终确定出8个候选化合物分子进行化学合成和后续湿实验验证。结果表明,8个分子中有3个分子的IC50抑制活性达到纳摩尔级别,其中1个分子不仅体内药效动力学和药代动力学(PK/PD)性质表现很好,而且同时兼有较长的半衰期及口服生物利用度,更重要的是,它相对于参考化合物分子具有更低血浆蛋白结合率。从新颖性来看,该系列化合物的分子骨架不在该靶点已知的被专利的化合物分子范畴之内,属于新颖的、有活性潜力的候选拮抗剂分子。目前,该系列分子已经申请专利保护(申请号:202210425594.5),相关研究成果即将见刊发表。
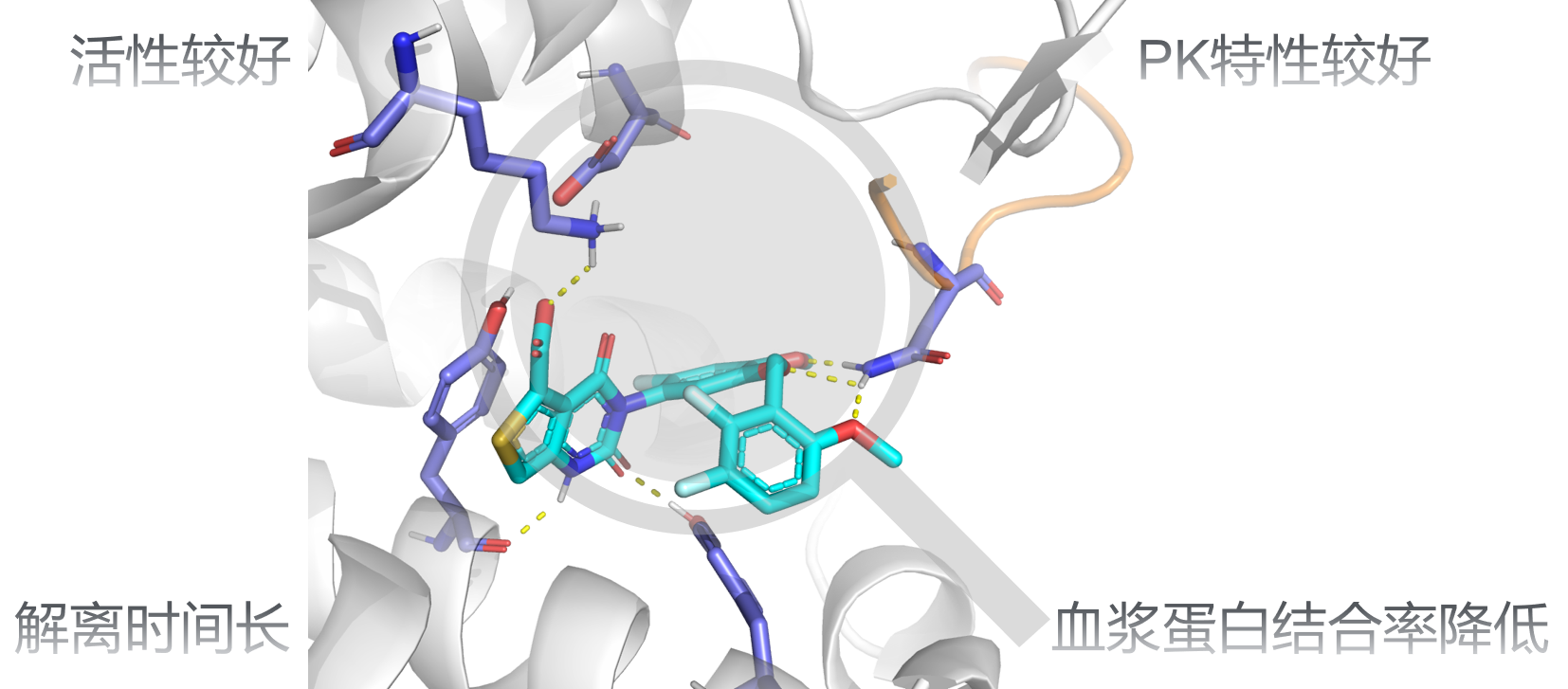
LS-MolGen能够生成拮抗效果好、半衰期较长、口服生物利用度较高并且血浆蛋白结合率更低的化合物分子
参考文献
- Deng, J. et al. (2022) Artificial intelligence in drug discovery: applications and techniques. Brief Bioinform 23 (1).
- Zhavoronkov, A. et al. (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 37 (9), 1038-1040.
- Skalic, M. et al. (2019) Shape-Based Generative Modeling for de Novo Drug Design. J Chem Inf Model 59 (3), 1205-1214.
- Ma, B. et al. (2021) Structure-Basedde Novo Molecular Generator Combined with Artificial Intelligence and Docking Simulations. J Chem Inf Model 61 (7), 3304-3313.
- Spiegel, J.O. and Durrant, J.D. (2020) AutoGrow4: an open-source genetic algorithm for de novo drug design and lead optimization. J Cheminformatics 12 (1).
