← Back
轻量型等变图神经网络助力蛋白质设计
2022年12月
近日,上海交通大学自然科学研究院王宇光团队与洪亮团队合作,设计等变图神经网络LGN,从蛋白质3D结构学习有益的蛋白突变。多点位突变任务中,在公共标准数据集CATH的预测水平超过Meta AI的SOTA大语言模型ESM,而计算效率提高10-100倍。初步结果以“Lightweight equivariant graph representation learning for protein engineering”发表在AI顶会《NeurIPS Workshop on Machine Learning in Structural Biology》上。
1. 研究背景
定向进化是抗体设计和酶工程等生物工程领域里的一个很重要的方法。它通过对蛋白质中特定的一个或多个氨基酸位点进行突变来优化野生型蛋白质的性质(比如热稳定性,发光强度,催化活性等),使其满足生物工程的需求。
一个蛋白质可能由成百上千个氨基酸组成,而每个氨基酸又有20种可能的类型。在传统的基于湿实验的生物研究方法中,构建有效的突变体需要在局部序列中进行贪婪搜索。为了获得具有更好性质的突变体,突变的点位还通常需要大于1个,也就是所谓的深度突变。然而由于深度突变的潜在突变组合总数是一个天文数字,巨大的实验成本使得对所有可能的突变体进行系统的实验测试成为一件十分困难、甚至不可能的任务。
另一方面,随着计算机科学、尤其是深度学习的不断发展与完善,很多虚拟预测与筛选方法在近几年都被提了出来,并在实践中得到了验证与广泛应用。然而,当前的方法大多是基于多序列比对(MSA)和/或蛋白质语言模型(PLM)来对蛋白质序列进行特征提取。前者提取出的蛋白质共进化信息的质量高度依赖同源信息的数量,比如在AlphaFold 2中就有研究表明一个蛋白质至少需要有30条以上的同源信息才可以得到比较可信的表示。但在实践中,并非所有蛋白质序列都是能进行同源比对的,例如,抗体互补可变域 (Shin et al. (2021) 的 CDR,也并非所有比对都足够深以训练足够大的模型以学习残基之间的复杂相互作用。另一方面,蛋白质语言模型来源于自然语言处理,因此模型主体通常需要搭建Transformer,递归神经网络,或其他自回归模型。这类方法通常需要海量的训练数据和复杂的模型设计,从而需要非常高的训练成本。即使是使用当前主流的预训练模型的思路,考虑到每个蛋白质都有独特的性质和进化方向,不涉及任何的重新训练路径而直接把一个预训练好的模型推广到任意的任务中,对于大模型的泛化性和表达能力也是一个巨大的挑战。
2. 论文概述
继续上述现状,我们设计了一个轻量级的深度几何预训练模型(lightweight graph network,LGN),结合蛋白质的结构信息对一条蛋白质上的每个氨基酸进行同步编码,用于设计合适的定向进化序列。
首先,由于蛋白质的序列决定了结构,结构又决定了功能,我们在输入数据时充分使用了蛋白质的结构信息。利用氨基酸序列的三维结构、每个氨基酸的物理和生化性质、以及它与周围不同尺度下的邻居氨基酸的相互关系,我们创立了蛋白质的图表示。在一个蛋白质图上,每个节点表示一个氨基酸,每个氨基酸根据k临近矩阵算法,与它在欧氏距离上最近的最多10个节点相连。我们还综合考虑了单一氨基酸的性质,比如它的残基类型,溶剂可及面积(SASA),B-factor等、前后相邻氨基酸的几何关系(比如二面角,本地坐标系等)、周围1阶邻居氨基酸的相互作用力、以及邻近氨基酸在蛋白质序列上的相对位置等,并利用这些不同尺度上的微观信息来定义图节点和边上的特征。
对于蛋白质的表征学习,我们使用具有旋转和平移等变性的图神经网络。根据物理学定律,无论蛋白质如何从一个地方平移或旋转到另一个地方,原子受到的力都应该保持不变。因此,为了尊重氨基酸的空间关系,也就是旋转和平移等变性,应将对称性的归纳偏差纳入基于蛋白质结构的模型设计中。一种直接的方式是类似图片处理类任务中常用的预处理方法,对输入数据进行增强。对于结构数据来说,另一种方式是针对性地设计等变神经网络,把蛋白质图的节点特征、连接方式、以及三维坐标都输入进模型,通过一系列具有等变性的信息传导网络层,对数据特征进行提取与更新。
另外,为了进一步利用生物学的先验信息来提高模型的泛化性和表达能力,我们还采取了三个额外的措施,包括:对输入的氨基酸类型进行加噪来模仿自然界中的随机突变;在氨基酸节点预测的损失函数打分机制中引入标签平滑来鼓励同类氨基酸之间的置换;利用多任务学习策略让预训练模型学习多种预测目标从而训练一个“一词多用”的图表示学习模型。

图:LGN模型框架
LGN的零样本学习训练框架如上图所示。首先,输入蛋白质数据集中的每个序列被k临近邻居算法转换成一个蛋白质图,并基于氨基酸性质提取出节点特征、边特征、以及氨基酸的三维坐标信息。接着,对一部分的节点特征进行噪声扰动后输入到等变图神经网络中学习图上的节点表示。这一节点表示被全连接层解码后可以预测多个不同的目标,比如去噪的节点氨基酸类型标签,SASA和B-factor数值等。这里的预测误差用于构建损失函数并传导回网络层进行反向传导。
在预测阶段,为了得到一个突变体的突变性质分数,我们首先把突变蛋白质的结构输入到预训练好的模型中得到每个节点的氨基酸类型概率。接着,我们把突变体和野生型蛋白质进行比对,并提取出不同部分的氨基酸、以及氨基酸类型对应的联合预测概率。这些数据进行对数处理、变换、加和后,结果的分数就是这个突变体的最终评分。
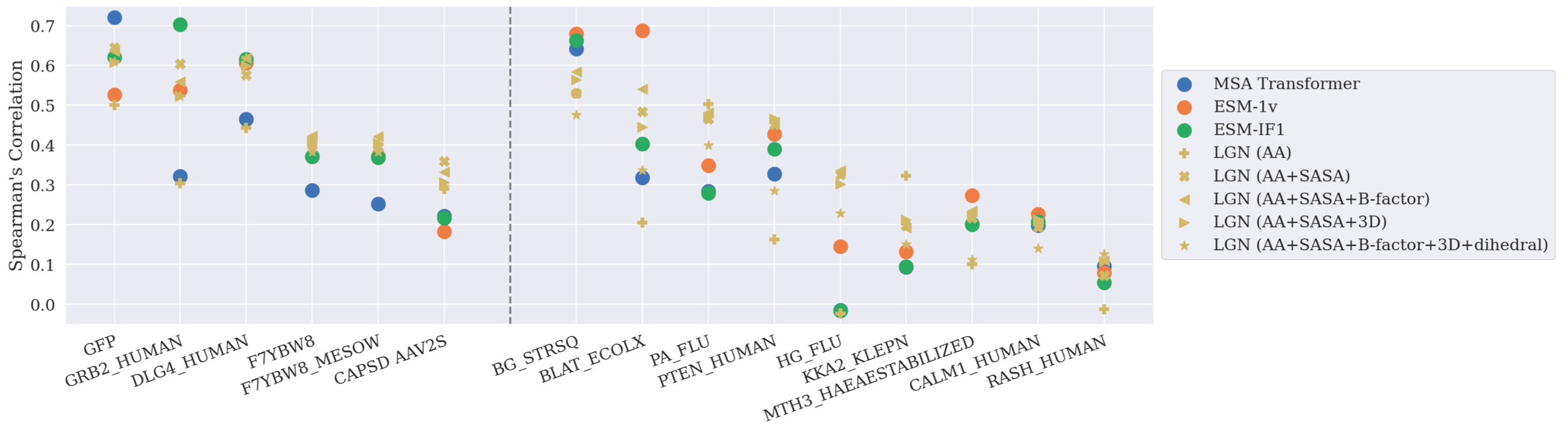
图:Per task Spearman’s correlation coefficients on the fitness of deep mutant effect prediction
为了检验LGN的预测效果,我们在15种不同蛋白质上进行了单点位和多点位突变的得分预测。整体来看,尤其是在多点位突变上我们超越了其他SOTA模型(如Meta AI的ESM)的结果。相比较于其他的对比方法,我们的模型参数和运行时间只需要百分之一。
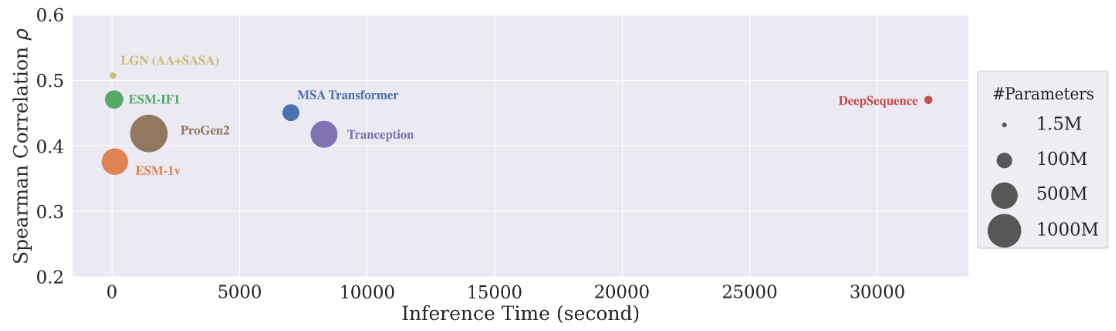
图:模型表现
3. 总结与未来方向
LGN的优秀表现来自于三个方面。
首先,通过多任务学习策略和生物学先验知识,LGN拥有很好的泛化能力。利用生物领域的专业知识,我们设计的预训练模型可以对给定氨基酸的微环境在不同尺度的化学和物理特性进行编码,从而获得更具有实际意义的表示。
其次,LGN对于每一个蛋白质可以一次性生成所有氨基酸类型的联合概率,从而避免独立突变假设。在计算高阶突变效应时,一种常见的做法是对相应的单点突变体的对数比值比(log-odds-ratio)进行加和。这种利用联合概率进行多点位突变打分预测的方式利用了定向进化中的上位性(epistatic effects),而在普通的独立突变中,对每个单独预测出的单点位突变进行线性组合很难保证这一性质。
第三,LGN在训练和推理阶段都有效地完成了相应的预测任务。通过把蛋白质的三维结构编码成具有拓扑性质的图输入,从而避免了二维序列或网格表示中通常需要的数据增强。同时,LGN在对由蛋白质图的几何结构定义的氨基酸微环境进行编码时,所使用的等变消息传递网络可以提供具有平移和旋转等变性的特征提取单元。
