← Back
SAM-DTA:全新策略的分子活性算法助力新药研发
2022年12月
近日,上海交通大学自然科学研究院/药学院洪亮教授课题组联合商汤科技,在基于深度学习的蛋白质小分子活性预测上取得重要突破。该工作采用全新的无需知晓蛋白质序列的策略,极大地提升了深度学习模型在活性预测方面的精度和计算速度,为基于人工智能的药物设计再添重要助力。该研究成果以《SAM-DTA: A Sequence-Agnostic Model for Drug-Target Binding Affinity Prediction》为题,已发表在深度学习主流杂志《Briefings In Bioinformatics》上。
1. 研究背景
分子在蛋白靶点上的活性是药物设计中的重要评价指标,因此分子活性预测也是深度学习在药物设计中的重要应用之一。一直以来,通过计算手段来预测分子活性是药物设计中非常常用的手段,从基于分子的QSAR模型,到基于结构的分子对接,都是分子活性预测的重要工具。随着近年来深度学习算法的发展以及大量分子活性数据的积累,采用深度学习来预测分子活性成为了近年来研究的热点。在之前大多数的分子活性预测模型中,蛋白质和分子会分别进行表征,虽然不同工作中对于蛋白质和小分子的具体表征方法存在一定差异,但是同时对蛋白序列和小分子建模的模式是一致的。在本工作中,作者打破这一固有模式,建立了无需知晓蛋白质序列的策略来预测分子和蛋白靶点的活性(sequence-agnostic model for drug–target binding affinity prediction, SAM-DTA)。该方法不显性的采用蛋白质表征,而是通过与其结合的小分子隐性地来表征,结合Multi-head联合训练策略,显著提高了预测精度和计算效率,在活性预测任务上达到SOTA效果,并且在全新蛋白的迁移学习场景和前瞻性数据的预测场景中也展现出极大的精度优势。
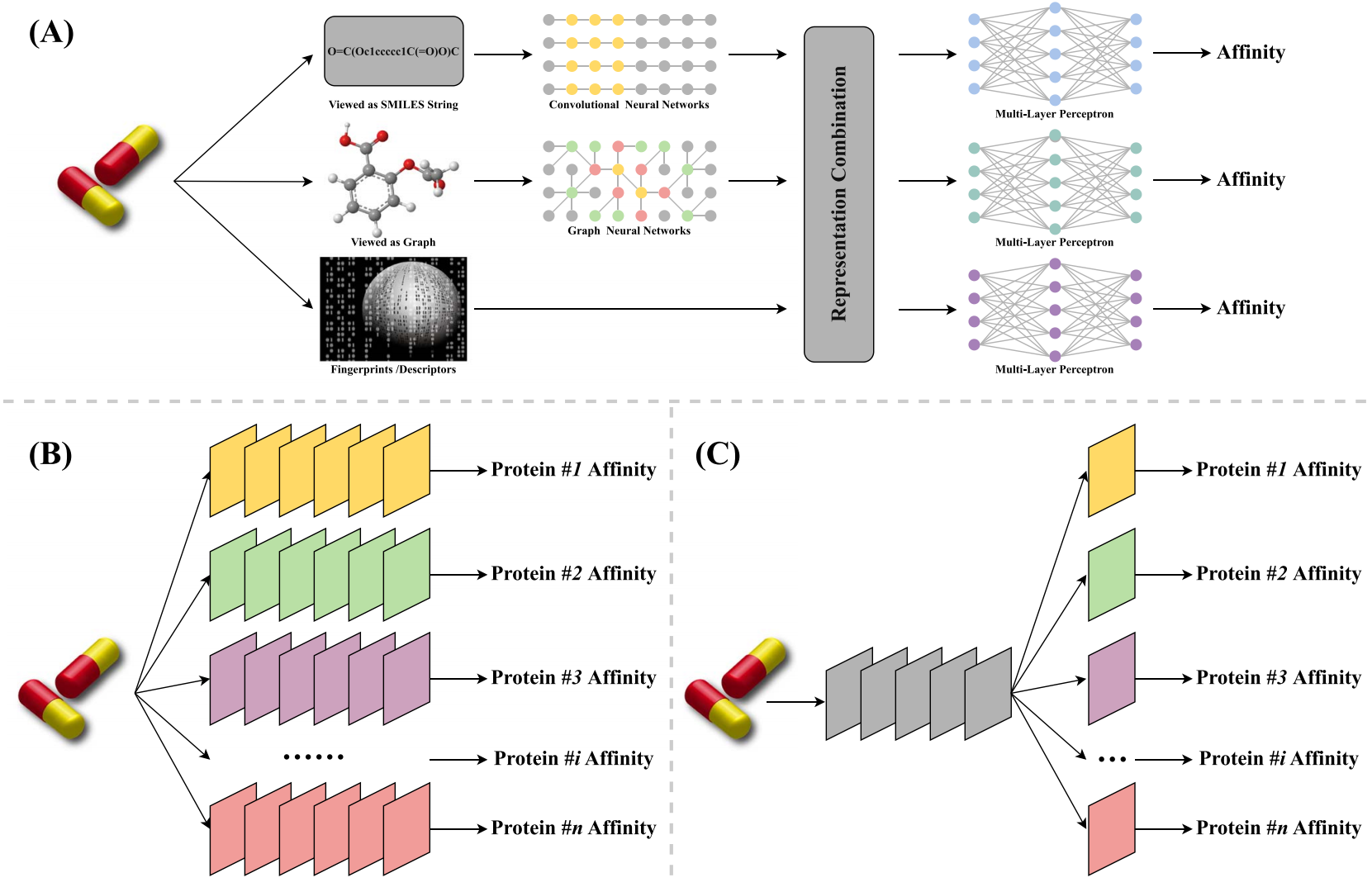
图1. SAM-DTA模型的整体概览图
2. 研究方法和数据集
2.1 数据集
模型的数据集来自于Karimi等人在2019年发表在《Bioinformatics》上的工作DeepAffinity。该工作中收集了截止到2018年BindingDB中的活性数据,通过过滤、去重得到了最终的数据集。此外,Karimi等人为了测试DeepAffinity的泛化性,将GPCR、激酶、离子通道蛋白、核雌激素受体这四类蛋白单独挑选出来作为额外的测试集。本工作中,为了满足模型的需求,我们选取了IC50数据,将这部分数据按照蛋白进行分类,去除活性数据少于200的蛋白。最终得到401个蛋白(291,504条活性数据)作为基本数据集BindingDB-18,上述四个不同类型蛋白的数据作为额外的数据集BindingDB-18ex(包含129个蛋白,91,767条活性数据)。
2.2 分子表征和网络架构
如背景中所提到,本工作中没有用到蛋白质的信息,因此对于模型来说只需要考虑小分子的表征。小分子表征一直以来是深度学习在小分子药物设计领域的研究热点,这里我们使用了传统的分子描述符和分子指纹,以及在深度学习常见的SMILES表征和分子图表征。同时,我们尝试了不同分子表征的组合,以达到最优的训练效果。此外,我们针对不同的表征采用了不同的网络架构,对于SMILES表征,我们采用了基于文本的RNN类的网络架构,以及CNN类的网络架构;对于分子图表征,我们采用了GNN的网络架构。此外,为了证明我们提出的SAM和Multi-head策略的有效性,我们分别设计了两个对照模型。对于前者,我们设计了蛋白质序列可知的Sequence-Aware模型;对于后者,我们设计了Singleton模型,将每个蛋白的数据分开单独训练。
3. 研究结果
3.1 SAM-DTA和其他模型的精度比较
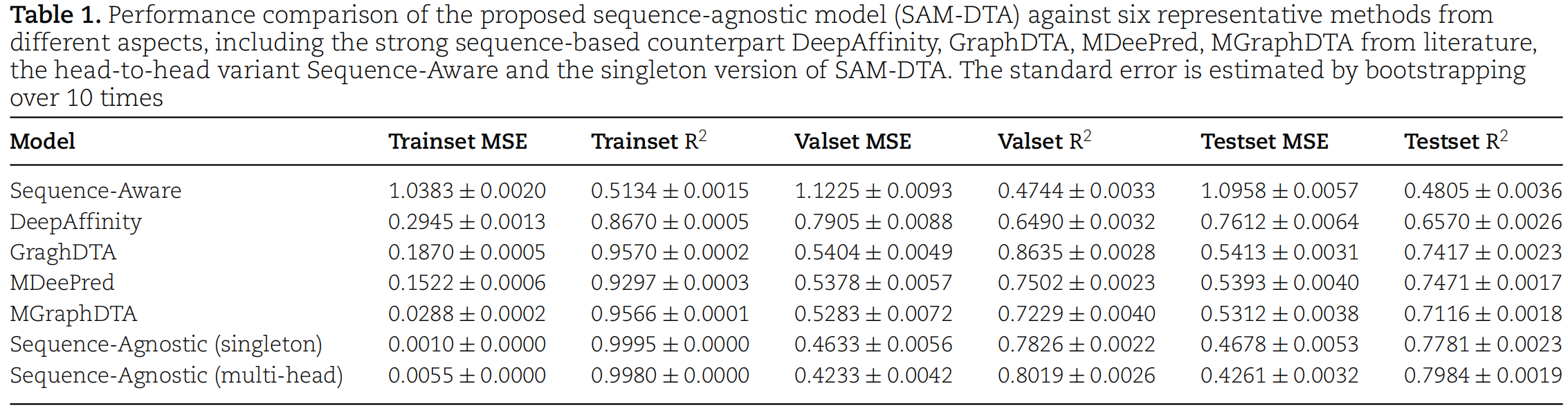
首先,我们将本工作提到的方法SAM-DTA与DeepAffinity、GraphDTA、MDeePred、MGraphDTA等其他相关的活性预测模型进行了对比,结果表明基于SAM策略的模型预测效果均明显优于其他模型,且基于Multi-head策略训练的模型效果更好。
表1. SAM-DTA与其他模型的精度对比
3.2 SAM-DTA使用不同分子表征和网络架构的精度表现
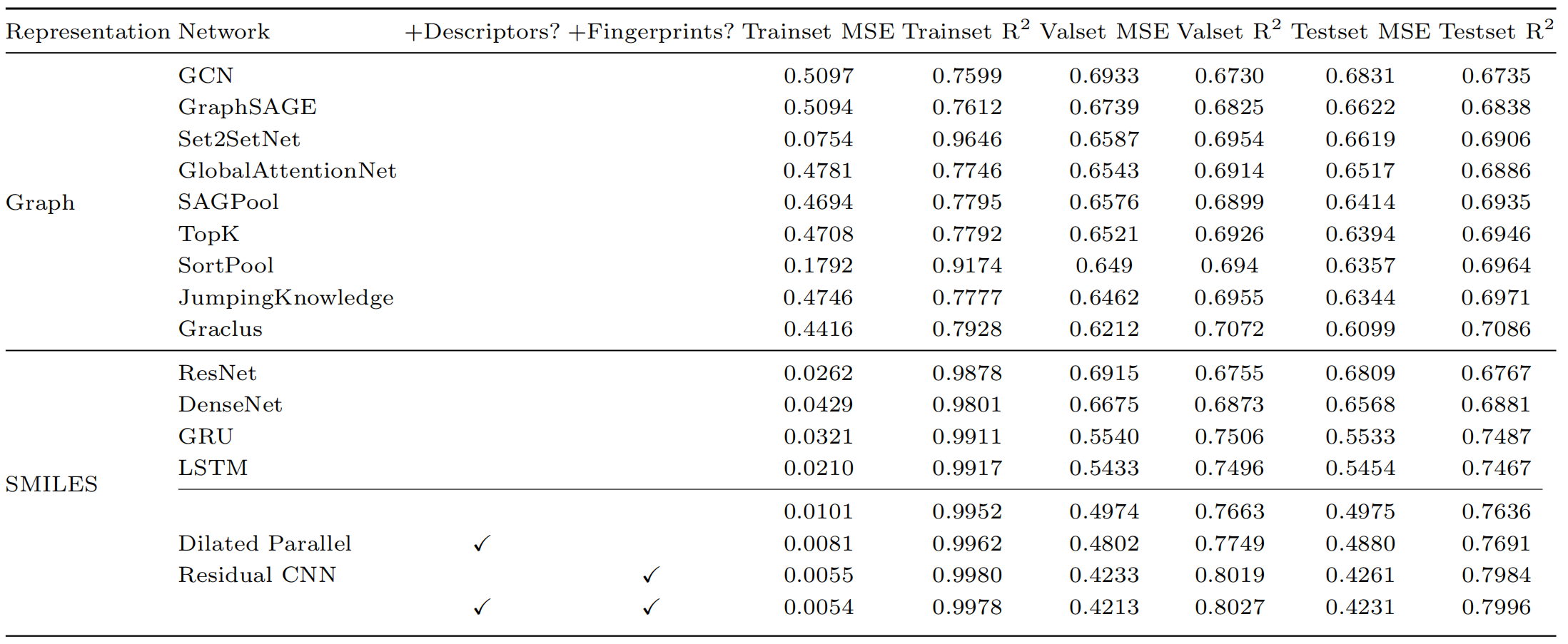
为了进一步找到适合SAM和Multi-head策略的分子表征和网络架构,我们测试了多种不同的分子表征,包括分子描述符、分子指纹、分子SMILES表征、分子图表征,以及多种不同类型的网络架构。结果如下表,当采用分子SMILES和CNN网络架构时结果更好,预测值和实验值的MSE较小,R2更大。此外,将分子描述符和指纹加入模型中,模型精度会更高。
表2. SAM-DTA在不同分子表征和网络架构下的精度
3.3 SAM-DTA在全新蛋白上的精度表现
上述场景都是对于已知蛋白的效果,那么SAM-DTA在全新蛋白上的表现怎么样呢?作者将方法中提到的BindingDB-18ex中的四类蛋白作为全新蛋白,然后采用以下两种方式来评估模型在全新蛋白上的效果。一种是将现有模型作为预训练模型,在全新蛋白质上微调,这也是深度学习中常用的策略;另一种就是从训练好的模型最后一层直接提取特征,然后用传统的机器学习方法进行训练。测试结果如表3所示,无论是前者还是后者,SAM-DTA的精度都要优于其他模型,而且SAM-DTA的优势在后者体现的更加明显。此外,RBF-SVR模型的精度和采用微调方式的精度相差不大,因此,如果在数据量较大的情况下,将SAM-DTA作为特征提取器,结合RBF-SVR在不损失精度的前提下可以大幅提高计算效率。
表3. SAM-DTA在全新蛋白的精度

3.4 前瞻性数据的研究
除了上述的测试外,该论文还测试了SAM-DTA对前瞻性数据的预测能力。如上文提到,SAM-DTA的训练数据和DeepAffinity工作基本一致,是截止到2018年BindingDB数据库里的数据。因此,我们新收集了2019年-2021年以及2019年-2022年的数据作为前瞻性数据。值得注意的是,2019年-2022年的数据量为232,186,数据量与训练数集基本一致,可见近三年来活性数据的增长速度之快,这也给模型带来了一定的挑战。因此,我们从两个方面来测试模型的能力,一方面将这部分数据直接当做测试集,另外一方面将该数据放入训练集中重新训练,比较模型在数据加入前后的精度。测试结果如下表所示,在第一项测试中,SAM-DTA的精度明显优于其他模型,虽然因新数据中存在一些极端情况导致预测精度有所下降,但是在大部分数据上效果与之前相同(如下图2所示)。在第二项测试中,将前瞻性数据加入训练集中,模型的精度会有大幅提升。该结果与预期一致,如果采用更多的数据来训练,模型的精度会有所提高。
表4. SAM-DTA在前瞻性数据上的精度

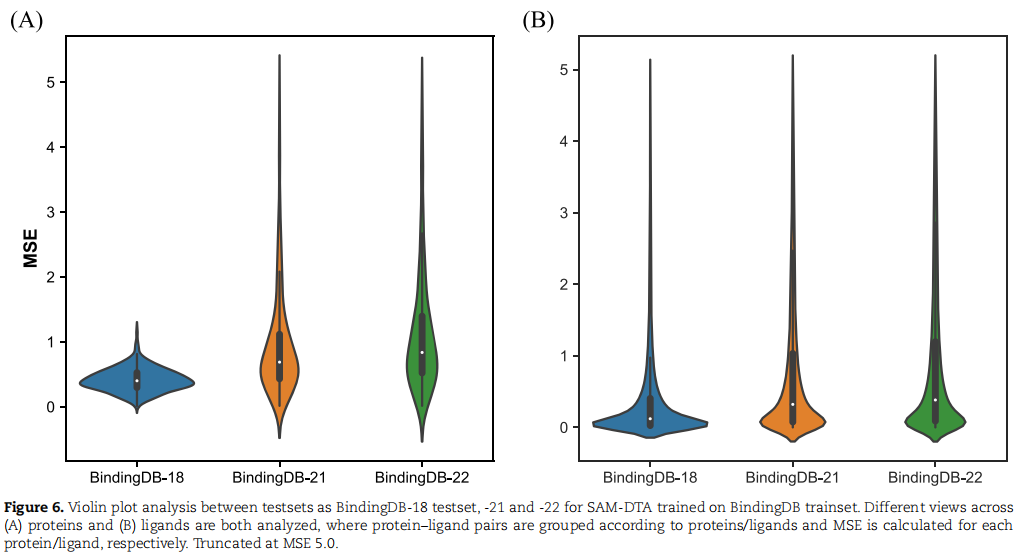
图2. SAM在不同测试集中预测值和实验值IC50的偏差(A)活性数据以蛋白分类的MSE分布(B)活性数据以小分子分类之后的MSE分布
4. 总结
在本工作中,作者提出了一种基于无需知晓蛋白质序列的策略活性预测模型SAM-DTA,与其他同时表征蛋白质和小分子的方法不同,该方法将不同的蛋白分而治之,然后通过Multi-head的方式进行联合训练。测试结果表明,SAM-DTA在常规活性预测情形和全新蛋白的迁移学习情形下都取得了SOTA效果,而且相比其他模型有较大的提升。此外,该模型还有很大的拓展性。从小的层面来讲,除了本工作中测试的IC50这个活性指标,SAM-DTA模型对于其他活性指标(例如Ki、Kd、EC50)同样适用。从大的层面来说,由于SAM-DTA序列不可知的模型,该方法未来在分子对肿瘤细胞、微生物干扰效果的预测任务同样适用。
本研究获得了国家自然科学基金(11974239、31630002),上海市教委创新计划项目(2019-01-07-00-02-E00076),上海人工智能实验室以及上海交通大学高性能计算和学生创新中心的支持。
